I think I talked free energy to death in the last two posts... Enough to be able to only need to mention ONE more time that it deals with which direction a reaction is going to be favorable in.
Reactions' abilities (or qualities?) of being
favorable are really fucking important in terms of biological processes because for shit to happen in your body (like protein synthesis and shit) it has to be somehow favored. Otherwise...it won't happen.
Sometimes, things that happen in our bodies aren't...favorable...Remember once again terms like endergonic and exergonic. Endergonic relates to things that are bad for -ΔG...so they deal with processes that are +ΔG which means the reverse of that reaction is actually what's favored. And it wouldn't really be good times if say... the reaction of "Amino Acids ----> Proteins" was going in reverse ALL THE TIME if it were +ΔG for it to go forward, because then we'd just be spontaneously turning into amino acids and our proteins would be soup. That wouldn't be good. Nothing would get anywhere.
But sometimes, that's what's up, and reactions that get shit done are endergonic. So what can we do?
These reactions, since they're like, necessary for life and shit, are going to be
coupled to reactions that ARE strongly favored in the forward direction.
Nice and BIG. So here we have two reactions, one which is ΔG+ (Thing ----> More Complicated thing) And one which is ΔG- (This ----> That)
If that's what's going on in the cell and the cell needs to do the first reaction, it can couple both the reactions together, adding up their ΔG's, and from there you get the cell converting
This Thing to
That Complicated Thing. That was totally unintentional when I was writing it on the board but it came out making sense-ish. And it's funny. To me.
In the body, the coupling of reactions is used to make things happen...bring things across membranes that membranes won't pass lazily, nerve impulse travellings, muscle contractions...
So what's up with all these phosphates, man?
For all this shit that's highly ΔG+, we need to make sure we have things that are ΔG- enough to make shit happen. For that, we have things called "transducers", which are basically things that convert stuff into slightly different stuff. These phosphates that we're going to talk about in a second are things that have all that highly negative free energy that's going to let other shit happen by coupling it to the stuff that happens with the phosphates. The phosphates undergo "hydrolytic release of phosphate groups" which basically means, in water, phosphate groups are released. The
hydrolysis of these compounds with phosphates has highly negative free energy, enough to allow other things to happen.
Oh god, what is this chart?
So, THESE are THOSE COMPOUNDS that have phosphates that are released in hydrolysis, which provides us with enough negative free energy to couple shitty ΔG+ reactions to. And on the next bage, there's a second, almost as intimidating table, except smaller and with less colors..Oh god, can we do this one big word at a time?
Hydrolysis of _____ Gives us ____kj/mol ? Will that help?
PEP -61.9 kj/mol
1,3-BPG -49.4 kj/mol
CP -43.1 kj/mol
ATP ... AMP (-45.6 kj/mol)
ATP ... ADP (-30.5 kj/mol)
PPi -19.2 kj/mol
(And the less exergonic ones...)
AMP -13.8 kj/mol
G-1-P -9.2 kj/mol
So I guess we can call Pyrophosphate our cutoff point for...exergonicity?
ATP is kind of really important so lets look at successive hydrolysis' of ATP and then the things that come out of it, which are also hydrolyzed with phosphates falling off all over the place.
So there's ATP. It's a Ribose ring (Note 2' OH group intact), with an Adenosine nitrogenous base, and three phosphate groups attached at the 5' end.
One thing we can pay attention to is which bonds are the phosphoanhydride bonds and which bond is the phosphate ester bond. Only one phosphate ester bond in ATP. Two phosphoanhydride bonds.
When you hydrolyze ATP, you get ADP (adenosine diphosphate) which is then hydrolyzed to AMP (adenosine monophosphate) which is then finally hydrolyzed to Adenosine by its sad lonely self.
With each round of hydrolysis, you get a Phosphate group released and some negative free energy being released as well. This is when a NEW ΔG term comes in (for this chart), which is ΔGs' (delta G standard-prime). This, also will be discussed, in a second.
Remember talking about K and Q and equilibrium constants and all that crap in the last post? Well....with the whole ATP thing, and the ΔGs' value being -30.5 kj/mol, we're discussing a reaction where the
equilibrium constant is greater than 10^5....which means that the equilibrium lies way far to the right, so far as to the point that we can consider this reaction
irreversible. Now, remember how in BIG SCARY TABLE, some things were really really exergonic while some things were less exergonic? Now we can talk about why. In organic chemistry 1, the big "joke" and also the serious
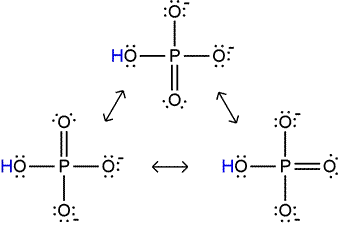
thing was that "The answer is always resonance". Well...let's talk about the resonance in this situation of figuring out why some things give us all this negative free energy when they are hydrolyzed and others...not so much.
Look at this thing that gets released, which is called
orthophosphate ion (HPO4 2-)
Look how stable that sucker is. All these versions of thing thing are of equal energy, which means
high entropy. Why does it have higher entropy? Remember, entropy likes randomness. When the Pi is bound up in an ester (like, before it gets released), it has
fewer possible resonance forms, which means it's less resonance stablizied (okay that's kind of redundant), but that means that when Pi is released from underneath it's mother's wing, there's more entropy, which is favorable.
Another factor as to figuring out the reasons behind the difference in exergonicity of stuff that's hydrolyzed and why some stuff has more free energy than other stuff is that the stuff that's the
products of hydrolysis can then ALSO be hydrolyzed. When the Pi gets released, especially if the other thing that's being release has a charge, that's good times for hydration land. IONS LIKE TO BE HYDRATED.
A third factor is backed up by us remembering that like repels like and opposites attract. Lets look at some of these phosphate-having compounds that get hydrolyzed and how there is repulsion between the
like charges that they have inside of them.
It might actually be in my best interests that this is a bit blurry since, there's bad words in it...But this just shows how in these compounds that get hydrolyzed, hydrolysis is favorable since it lets all these negative charges stay the fuck away from each other. The charge-charge repulsion forces are strong with these ones.
The next thing deals with keto-enol tautomerization...One of the things we learned about in Orgo 2, happening to be one of the questions I blew on the first Orgo2 exam and then later when Lenny yelled at us, it was really drilled into my head to understand what the fuck is going on. Because rule #1 is to LISTEN TO WHAT LENNY SAYS or else you're going to fail life and not be a doctor. So yeah...keto-enol, I think I can draw that shit in my sleep now.

In the little box on the left I drew the typical example of a keto-enol tautomerism, and now let's talk about what the fuck the deal is with phosphoenol pyruvate and why the fuck the ΔGs' for this thing is a whopping -61.9kj/mol...Well THEN. When PEP is hydrolyzed, what's up is that there's a keto-enol tautomerization that occurs in the product. Since the DIRECT product is in the enol pyruvate form, it quickly tautomerizes to the keto form of pyruvate, which is thermodynamically favored. The extra ΔGs' negative-ness basically swarms in from the goodness that comes out of the tautomerization.
Another thing we see in these hydrolysis reactions that release Pi when the compounds are hydrolyzed is that an H+ is released. This is where I guess that second less scary chart comes in (didn't take a picture of it, but we're gonna talk about it now) where they bring in how pH factors into some of these compounds, not others.

Here's what's good with ATP hydrolysis, and the ΔG that we can calculate from it. We usually deal with reactions that are around neutral pH so we need to talk about the chemical potentials (measure of how much that thing contributes to ΔG) of the water and the H+ in a different way than we did before. In the previous situations, with biochemical "standard state" stuff, the activity of water is not affected by any reactions that are going to use it up or produce it...another way of saying this is that in the biochemical standard state stuff, the "activity" of water is "unity". While standard state concentration is 1 M solutions, in our bodies, the concentration of H+ is 10^-7 which is a LOT less than 1M. So now we need to start looking at the H+'s contribution to ΔG as a "thing" that is found at 10^-7 M and not as something that's going to be a standard 1M concentration.
So in this new state that we're talking about (living shit state), 1M is not going to be considered unity for H+. We're now going to call H+'s activity unity when the concentration of H+ is 10^-7 M. THIS is where this millionth new way of talking about ΔG (this time, we add two things to it, to make it ΔGs') comes in. And now we can plug some pseudostuff into that equation:
Now, we need more numbers, make stuff more real for us. You can tell that this stuff is real because it has NUMBERS and an ANSWER which is what "matters in life". Having answers. Which, we won't always have, in life.
Another important thing when we look at that big scary table of phosphate-containing compounds and how those compounds can be hydrolyzed and release a phosphate, is that some of them have something called phosphate transfer potential which is important. Phosphate transfer potential havingness means that some compounds can phosphorylate others. This potential is defined as -ΔGs'. So looking at bigscarytable again...

See that blue thing on the right that is labeled TRANSFER POTENTIAL? And it even has a SCALE that tells you what's good? Well our good friend PEP is at the top of the chart..remember this is the thing that gets all its bragging rights from keto-enol tautomerization....So, PEP is at the top of the list, with the highest transfer potential, and that sad G-1-P is at the bottom of the list with a weaksauce transfer potential. Things that are above things can drive the phosphorylation of anything below them as long as you can couple stuff. Coupling is done by making the reaction occur on the surfaces of protein molecules, like enzymes. Let's look at these coupling reactions for this stuff:
Hydrolysis of PEP is ΔGs' = -62 kj/mol
Phosphorylation of ADP = +30. kj/mol
So when you couple, phosphorylation of ADP by PEP is going to be ΔGs' = -31.4 kj/mol
PEP has more phosphate transfer potential than ATP, SO THAT MEANS it can give ADP a phosphate group and it's going to be thermodynamically favored and YOU'RE GOING TO LIKE IT. Likewise, ATP can give glucose a phosphate group (I'm calling phosphorylation "giving") because the phosphate transfer potential of ATP is greater than that of G6P:
ATP Hydrolysis is ΔGs' -30.5 kj/mol
Glucose Phosphorylation is ΔGs' +13.8 kj/mol
So when you couple, the phosphorylation of glucose by ATP is ΔGs' = -16.7 kj/mol
Finally, the chapter 3 ends off by talking about ΔGs' for REDOX reactions and I'm going to make the last chapter 3 post discussing that shit.


.JPG)